Our laboratory studies the pathophysiology of acute lung injury and how to promote recovery or prevent aberrant recovery from lung injury to prevent development of fibrosis. Acute respiratory distress syndrome is a severe hypoxemic respiratory failure that commonly develops after alveolar structures are damaged by pneumonia, for example from SARS-CoV-2 or influenza virus infection. Despite recent advances in understanding its pathophysiology, therapies aimed at reducing lung injury have been relatively ineffective, and its case fatality remains high (30%–45%). One alternative approach is to promote lung regeneration by stimulating stem/progenitor cells to replace damaged lung epithelium and to prevent aberrant repair resulting in fibrosis. Several epithelial subpopulations can serve as stem/progenitor cells in the reparative process after lung injury, yet little is understood about what intrinsic factors and microenvironment affect the alveolar epithelial stem/progenitor cells and their differentiation.
We aim to determine how mitochondrial metabolism regulates lung stem/progenitor cells as it relates to lung repair and injury, and to identify translational targets for disease monitoring and therapy. We are particularly interested in how lung epithelial cells repair after injury and what factors lead to aberrant repair resulting fibrosis. Mitochondrial dysfunction is commonly observed in patients with severe pneumonia and/or pulmonary fibrosis. By applying a multidisciplinary strategy with elaborately designed genetic knock-out and knock-in mouse systems, in vitro organoid models, RNA sequencing, and metabolomics, we are testing whether mitochondrial function is necessary for proper lung epithelial stem/progenitor cell differentiation. Findings from these studies can be directly applied to patients admitted to our hospital with pneumonia, influenza, and SARS-CoV-2 infection.
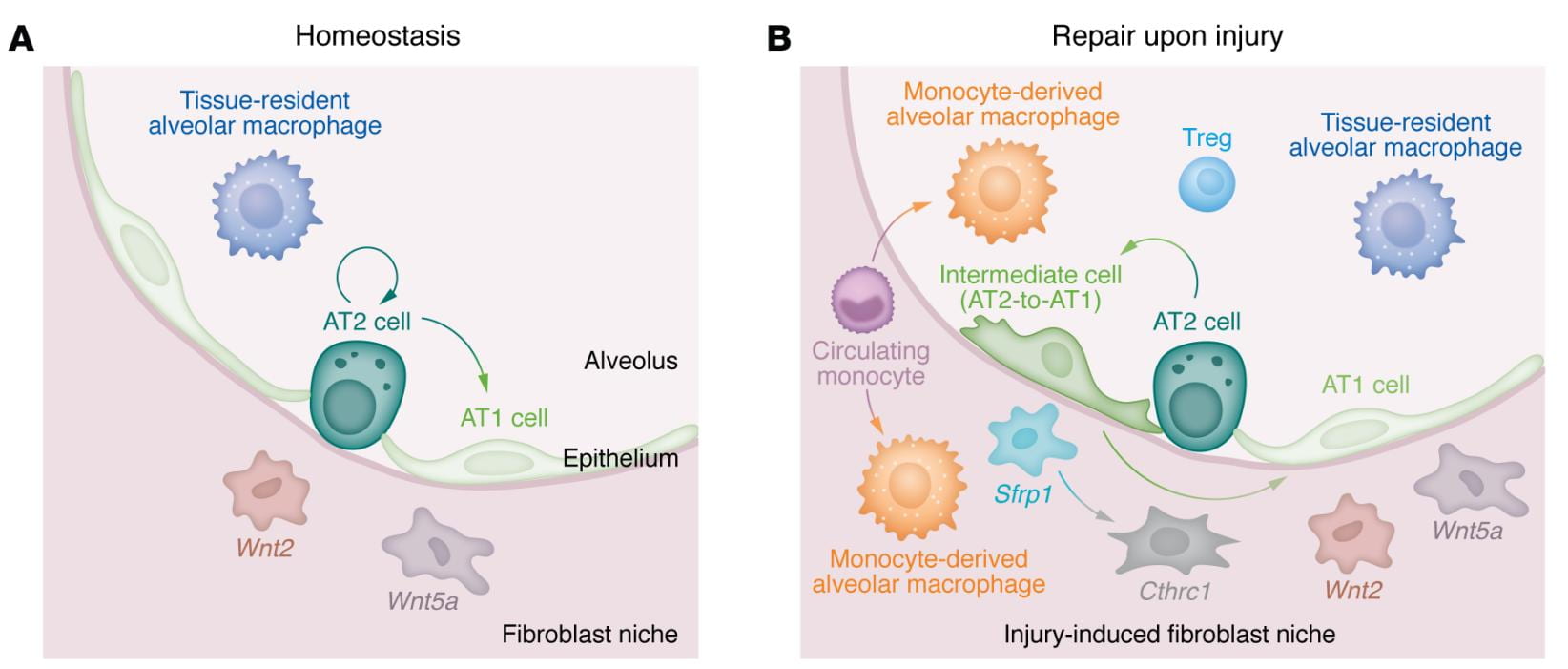
Repair of alveolar epithelium after lung injury
Alveolar epithelial type 2 (AT2) cells are small cuboidal cells that serve as a partially committed progenitor population. AT2 cells are required for the differentiation and maintenance of tissue-resident alveolar macrophages and are maintained by signals from adjacent mesenchymal cells, including Wnt2+ fibroblasts. Alveolar epithelial type 1 (AT1) cells are large flat cells that can spread over more than one alveolus. Their differentiation and maintenance in the niche also require signals from the adjacent mesenchyme, including Wnt5a+ fibroblasts.
In response to injury, AT2 cells undergo asymmetric division, with the smaller daughter cell regenerating the AT2 cell and the larger daughter cell differentiating into an AT1 cell. Single-cell RNA sequencing of the lung in murine models of injury, repair, and fibrosis and patients with pulmonary fibrosis identified a population of transitional cells with intermediate phenotypes between AT1 and AT2 cells that accumulate in areas of fibrosis.
Transitional cells and profibrotic monocyte-derived alveolar macrophages recruited in response to epithelial injury drive the differentiation of fibroblasts into an intermediate phenotype characterized by expression of Sfrp1 and subsequently a myofibroblast, characterized by Cthrc1. Regulatory T cells (Tregs) provide signals that directly or indirectly enhance epithelial repair.
This diagram is from our review on alveolar epithelial regeneration in the aging lung published in The Journal of Clinical Investigation.
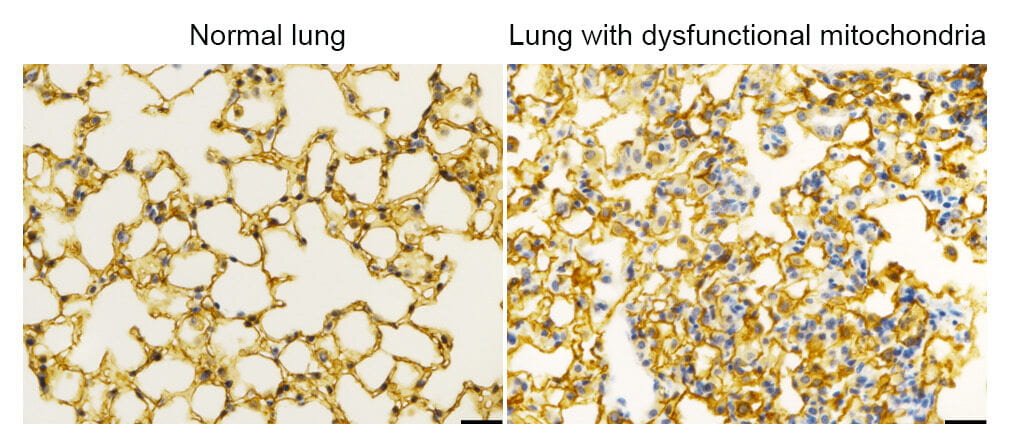
Dysfunctional mitochondria affect alveolar structure
We found that deleting the mitochondrial electron transport chain complex I subunit Ndufs2 in lung epithelial cells during mouse gestation led to death during postnatal alveolar development. Affected mice displayed hypertrophic transitional cells with both AT2 and AT1 cell features.
This image shows immunohistochemistry for AT1 marker podoplanin in normal murine lung tissue (left) and lung tissue from an Ndufs2 conditional knock-out mouse (right). The aberrant alveolar structures in the conditional knock-out mouse demonstrate that functional mitochondria are required for normal lung stem/progenitor cell differentiation.
Single-cell RNA sequencing revealed enrichment of integrated stress response (ISR) genes in transitional cells. We are currently examining the possibility that mitochondrial ISR signaling could be disrupted during lung repair after injury, resulting in prolonged viral pneumonia or pulmonary fibrosis.
Our findings, published in Nature, were reported by the Feinberg News Center and mentioned in the announcements of record-breaking research funding at both Feinberg School of Medicine and Northwestern University in fiscal year 2023.