Research Themes
Charge Transport in Disordered Materials
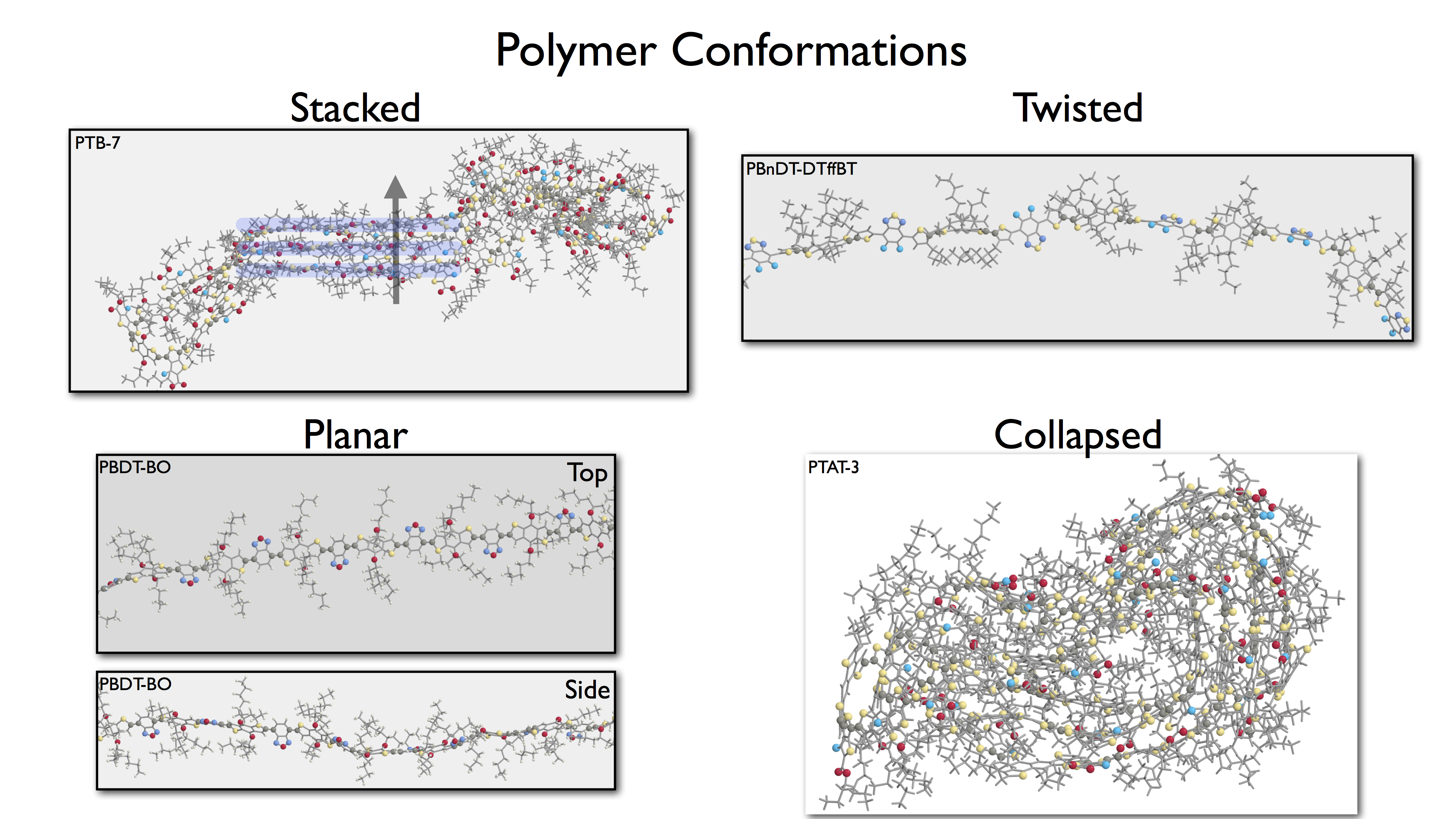
Donor-acceptor polymer morphologies can be predicted using molecular dynamics simulations provided an appropriate force-field has been developed for these polymers, where long-range dispersion plays a key role in determining the structural properties. Together with electronic structure calculations, we have developed a donor-acceptor molecular dynamics force-field that predicts many of the features seen in absorbance spectrum of the polymers in the melt state.
Charge Transport Mechanisms

Photovoltaic Design via Machine Learning
Sustainable progress in organic semiconductor design ultimately requires a quantitative predictive understanding of the way materials transport properties respond to structural disorder, a fact which is especially true for solution-processed organic photovoltaics. Combining molecular dynamics, electronic structure, and network analysis I am interested in mesoscopic transport networks that emerge commonly used photovoltaic charge acceptors (fullerenes).

Structural Dynamics of Disordered Proteins
We develop MD tools to investigate the structural dynamics of large biomolecules using environmental perturbations like T-jump, pH-jump, and oxidation/reduction of coordinated metal. Since structural dynamics away from the native state usually involves disordered states, sampling techniques such as maximum entropy classification and parallel replica exchange are necessary to predict functionally relevant structures. Given most of the experimental variables can only be estimated, we then deploy the MD methodology to refine the experimental factors of the environmental perturbations based on a Bayesian protocol.

Plasmonic Coupling of Self-Assembled Nanostructures
Anisotropic nanostructures can be self-assembled into 1-D lamellar structures using DNA hybridization. These structures show unique plasmonic characteristics.
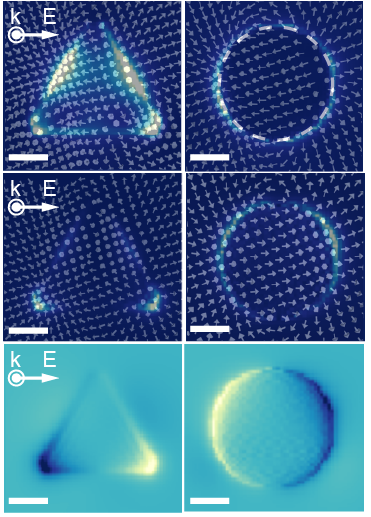

The calculation of LSPR spectra for assembled anisotropic metallic clusters requires knowledge of not only the spatial distances between clusters, but also their orientational behavior. Orientational symmetry breaking leads to emergent quadrupole and hexapole resonances due to the pi-coupling of the resonanances.
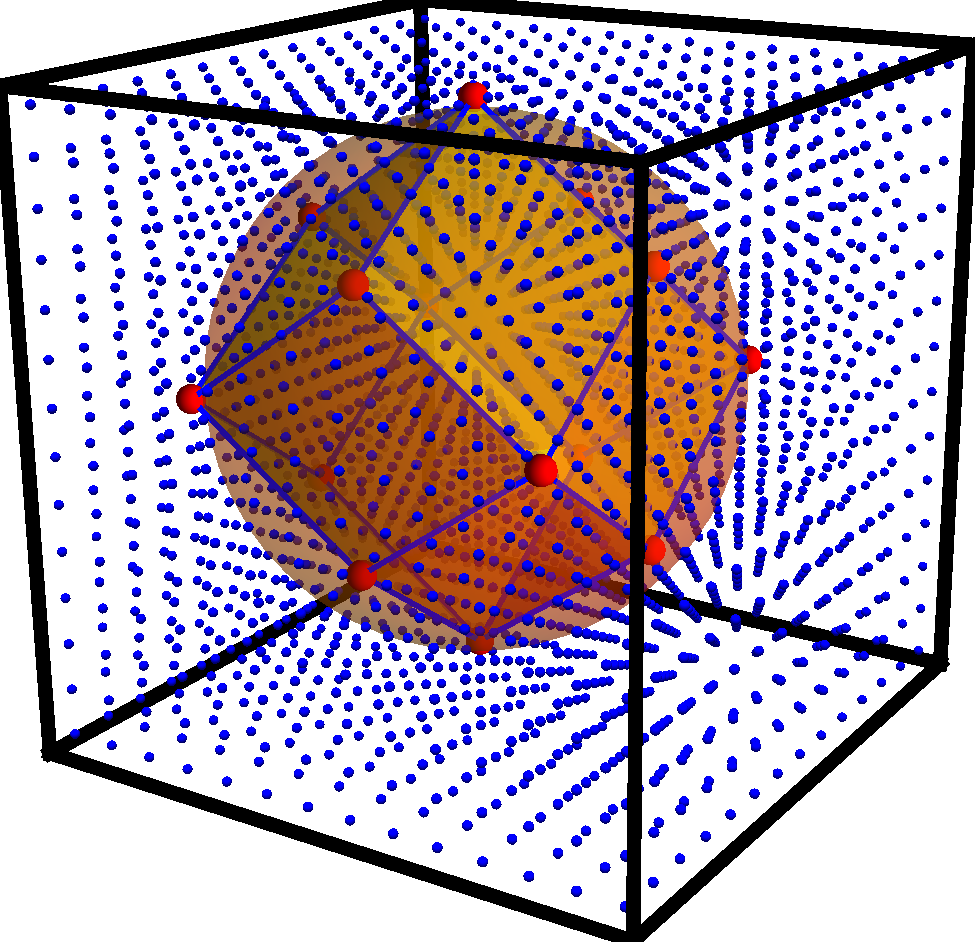
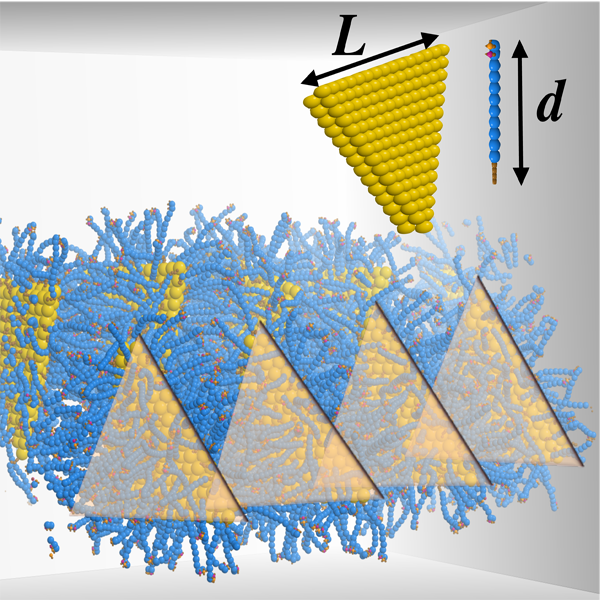
Self-Assembly of Anisotropic Colloids
The recent control in nanostructure shape in synthesis techniques allows simulators to predict assembly phase diagrams, optical properties, and surface functionalization as a function of shape. New shape libraries are needed for the different simulation suites such as molecular dynamics, electrodynamics calculations, and electronic structure calculations.
I am developing tools to create nanostructure models of a wide variety of shapes and symmetries. The model structures can further be adjusted to match details provided by electron micrographs with image libraries. I am actively developing the code in python to generate these structures.