Current Projects
- Molecular and Cellular Mechanisms of ACE1 mutations in Alzheimer’s Disease
The Cure Alzheimer’s Fund’s Alzheimer’s Genome Project identified a missense mutation in the angiotensin converting enzyme 1 (ACE1) gene that is a novel Alzheimer’s disease risk variant. ACE1 is best known for its role in blood pressure control, and hypertension has been previously associated with AD. However, the substrates and physiological functions of ACE1 are complex, and the mechanism by which ACE1 mutations are involved in AD pathogenesis is unknown. 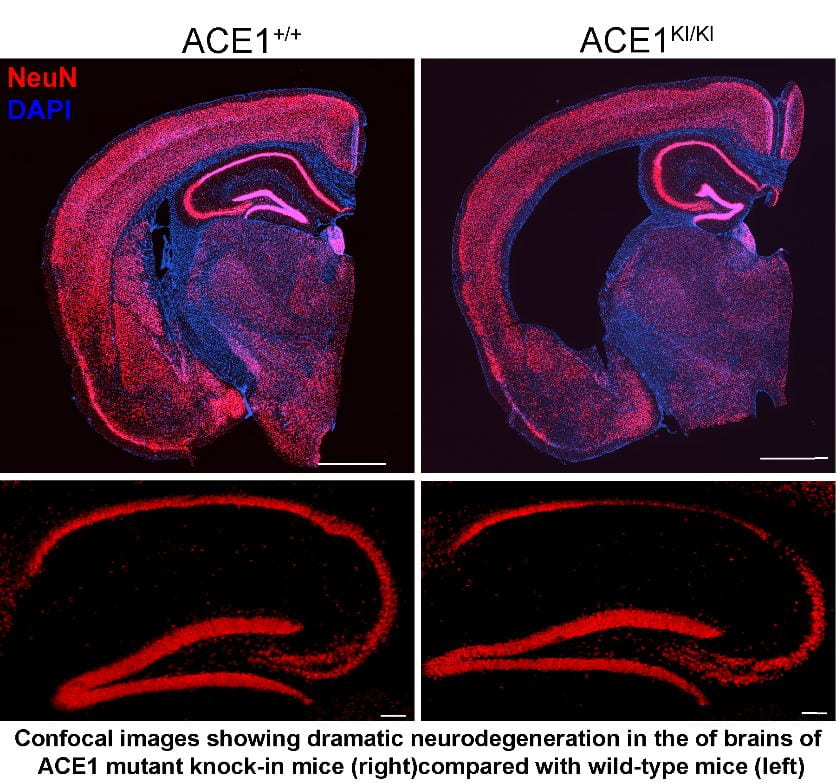 To gain insight into the role of ACE1 in AD pathophysiology, CAF’s Genes to Therapies program commissioned Taconic to make knock-in mice using CRISPR/Cas9 with the cognate mutation in the murine Ace1 gene. We have investigated the effects of the AD-associated ACE1 mutation in stable SH-SY5Y cell lines, ACE1 KI primary neurons, and cohorts of aged ACE1 knock-in and wild-type mice. Unexpectedly, we found that the ACE1 mutation causes cell death and neurodegeneration. Clinically available ACE1 inhibitors and angiotensin II receptor blockers prevented neurodegeneration in knock-in mice. Moreover, Aβ42 appears to exacerbate the intrinsic property of the ACE1 mutation to cause neurodegeneration. We are working to further understand the role of the ACE1 mutation AD pathogenesis.
To gain insight into the role of ACE1 in AD pathophysiology, CAF’s Genes to Therapies program commissioned Taconic to make knock-in mice using CRISPR/Cas9 with the cognate mutation in the murine Ace1 gene. We have investigated the effects of the AD-associated ACE1 mutation in stable SH-SY5Y cell lines, ACE1 KI primary neurons, and cohorts of aged ACE1 knock-in and wild-type mice. Unexpectedly, we found that the ACE1 mutation causes cell death and neurodegeneration. Clinically available ACE1 inhibitors and angiotensin II receptor blockers prevented neurodegeneration in knock-in mice. Moreover, Aβ42 appears to exacerbate the intrinsic property of the ACE1 mutation to cause neurodegeneration. We are working to further understand the role of the ACE1 mutation AD pathogenesis.
- Molecular and Cellular Mechanisms of the UNC5C Netrin Receptor in Alzheimer’s Disease Pathogenesis
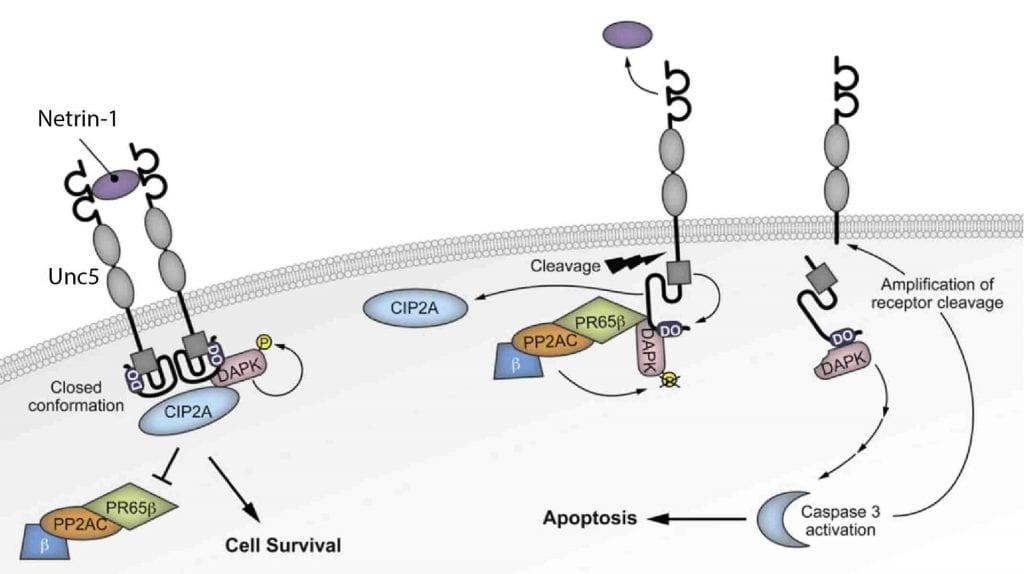
UNC5C is a netrin receptor in which a rare coding mutation, T835M, was recently identified that predisposes to late onset (LO) Alzheimer’s disease (AD). Our preliminary suggest that UNC5C T835M causes neuron loss in the presence of Aβ pathology. We hypothesize that UNC5C T835M predisposes to LOAD by making neurons more vulnerable to cell death induced by pathogenic Aβ and Tau. We will 1) define cell death pathways in 5XFAD and P301S-Tau mouse models of Aβ and Tau pathology, respectively, crossed to knockin (KI) mice in which UNC5C T835M is expressed endogenously; 2) identify cell death pathways in human induced pluripotent stem cell (hiPSC) neurons generated from UNC5C T835M patient fibroblasts; 3) use mass spectrometry (MS)-based proteomics to determine the comprehensive UNC5C T835M cell death proteome. This work is expected to provide proof of concept for therapeutic targeting of the UNC5C pathway for prevention of neuron loss in AD.
- BACE1 as a Therapeutic Target for Alzheimer’s Disease

b-Site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) initiates the formation of b-amyloid (Ab) that has a critical role in Alzheimer’s disease (AD). BACE1 is a prime AD therapeutic target and small molecule BACE1 inhibitors are in clinical trials. However, the safety and efficacy of BACE1 inhibitors have been challenged, because several BACE1 inhibitor trials have been terminated due to toxicity or lack of efficacy. BACE1 inhibitors may cause side effects, because BACE1 has many substrates with functions in neurons. The recent trial terminations suggest AD patients with dementia may be too late for clinical benefit from BACE1 inhibitors. These drugs may achieve greater efficacy at an earlier AD stage in presymptomatic individuals, so BACE1 inhibitor prevention trials are being planned. However, three key questions regarding BACE1 inhibitors for AD prevention remain unanswered. First, what level of long-term BACE1 inhibition is safe? Second, will a safe level of BACE1 inhibition slow AD pathology? Third, what Ab load is too high for efficacy of BACE1 inhibition? To answer these questions, we will use adult BACE1 and BACE2 conditional knockout (cKO) mice and treatment with the BACE1 inhibitor verubecestat we will determine the thresholds of BACE1 inhibition toxicities. Comparison of genetic and pharmacologic experiments will allow us to differentiate on- vs. off-target effects of BACE1 inhibition. Using amyloid plaque time-stamp analysis and other methods we will study the PDAPP amyloid mouse model crossed to adult BACE1 cKO mice or treated with verubecestat to determine a safe level of BACE1 inhibition that also slows the progression of Ab pathology. To identify the Ab load beyond which BACE1 inhibition may have little effect on tau pathology and AD progression, we will perform BACE1 cKO and verubecestat experiments on PDAPP mice with different Ab loads and analyze Ab and pathologic phospho-tau markers by biochemistry and histology. These experiments will generate translational information that will be valuable for BACE1 inhibitor clinical trials for the prevention of AD.
- Development of sAPPβ and sAPPα as Biomarkers of BACE1 Activity: Implications for Differentiation of Alzheimer’s Disease Subgroups

Cerebral accumulation of amyloid-beta (Aβ) peptides has a critical early role in Alzheimer’s disease pathogenesis. Cleavage of Amyloid Precursor Protein (APP) by β-secretase (also called BACE1) produces the sAPPβ fragment and this event must occur before Aβ may be subsequently released from the APP C-terminal fragment. Although BACE1 levels are increased in Alzheimer’s brain, it is unclear whether BACE1 elevation leads to increased APP cleavage and Aβ production in the human CNS. We employ the novel, highly sensitive method of stable isotope labeling, combined with immunoprecipitation/liquid chromatography/mass spectrometry, to quantitate kinetics of BACE1 activity in vivo. Measuring sAPPβ and sAPPα kinetic rates, we propose to determine if and by how much BACE1 activity is increased in late-onset AD (LOAD) subjects and in what subgroups of LOAD the shift toward increased BACE1 processing of APP occurs. Our preliminary results suggest that newly generated sAPPβ is increased in the cerebrospinal fluid of amyloid-positive Alzheimer’s subjects. We hypothesize that a subgroup of the LOAD and non-demented Amyloid+ populations overproduce Aβ because of increased BACE1 activity. We anticipate that kinetic rates of sAPPβ and sAPPα as biomarkers of BACE1 activity, together with kinetic rates of Aβ isoforms, will allow differentiation of AD subgroups based on different pathogenic mechanisms.
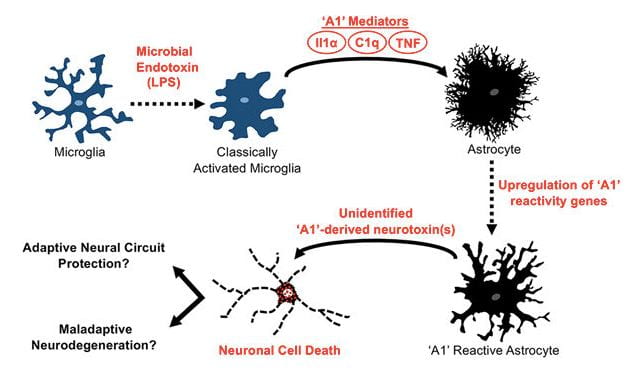
- Interaction of the microbiome with astrocytes and amyloid pathology
Cerebral accumulation of the Ab peptide in amyloid plaques is an early pathological hallmark of AD and likely triggers neuroinflammation and tau pathology in the disease1. Understanding the cause Ab accumulation and its effects in the brain are necessary for discovery of AD diagnostic biomarkers and disease-modifying therapies. Ab is generated via sequential proteolysis of the amyloid precursor protein (APP) by the enzymes b-secretase (BACE1) and g-secretase. BACE1 is the rate-limiting enzyme for Ab production. APP and BACE1 are increased by physiological stress and injury conditions that exacerbate amyloid pathology. Astrocytes are part of the innate immune system and are strongly affected by microglial cells. Indeed, activated microglia release proinflammatory cytokines that induce the A1 subtype of neurotoxic reactive astrocyte. However, the role of astrocytes, their interaction with microglia, and the effect of the microbiome on astrocytes in AD are poorly understood. Based on Dr. Sisodia’s and Tanzi’s work, we hypothesize that pathogenic microbiota in the gut and brain lead to elevated numbers of A1 astrocytes, perhaps via induction by microglia, resulting in negative consequences like neurotoxicity, increased levels of astrocytic APP, BACE1 and Ab generation, or decreased Ab clearance. The gut microbiome may signal to microglia in the brain via blood-borne cytokines, while the brain microbiome may directly interact with microglia and astrocytes. These processes could be affected by ApoE, TREM2, CD33, and other AD risk genes or a dysfunctional blood-brain barrier. We will investigate how the microbiota affect the astrocyte transcriptome, translatome, and metabolome in ways that could be important for AD pathogenesis.