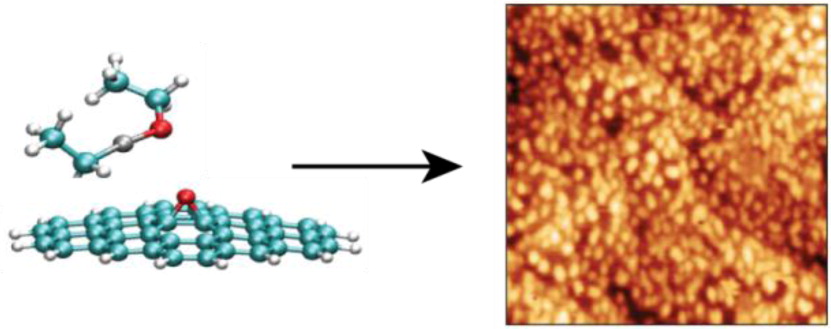
Above: Atomic oxygen produces epoxide groups on graphene, which act as reactive nucleation sites for zinc oxide nanoparticle growth using the atomic layer deposition precursor diethyl zinc.

Molecular dynamics simulations can be classified depending on how the potential energy of the system is calculated. One way to calculate the energy is to use wavefunction or density functional theories to solve the electronic Schrödinger equation. The most accurate electronic structure methods are extremely expensive from a computational standpoint and are only feasible for small system sizes (<100 atoms). The second way to calculate the energy of a system is by using classical force fields in which simple potential energy functions (e.g. Coulomb’s Law) are parametrized to specifically reproduce electronic structure or experimental results. The advantage of such potential energy surfaces is systems with up to a million atoms can be studied possibly up to the microsecond time scale. The main disadvantages of these simulations is the energies are not as accurate as electronic structure methods and the molecular topology is often predetermined at the beginning of the simulation so that chemical reactions do not take place.
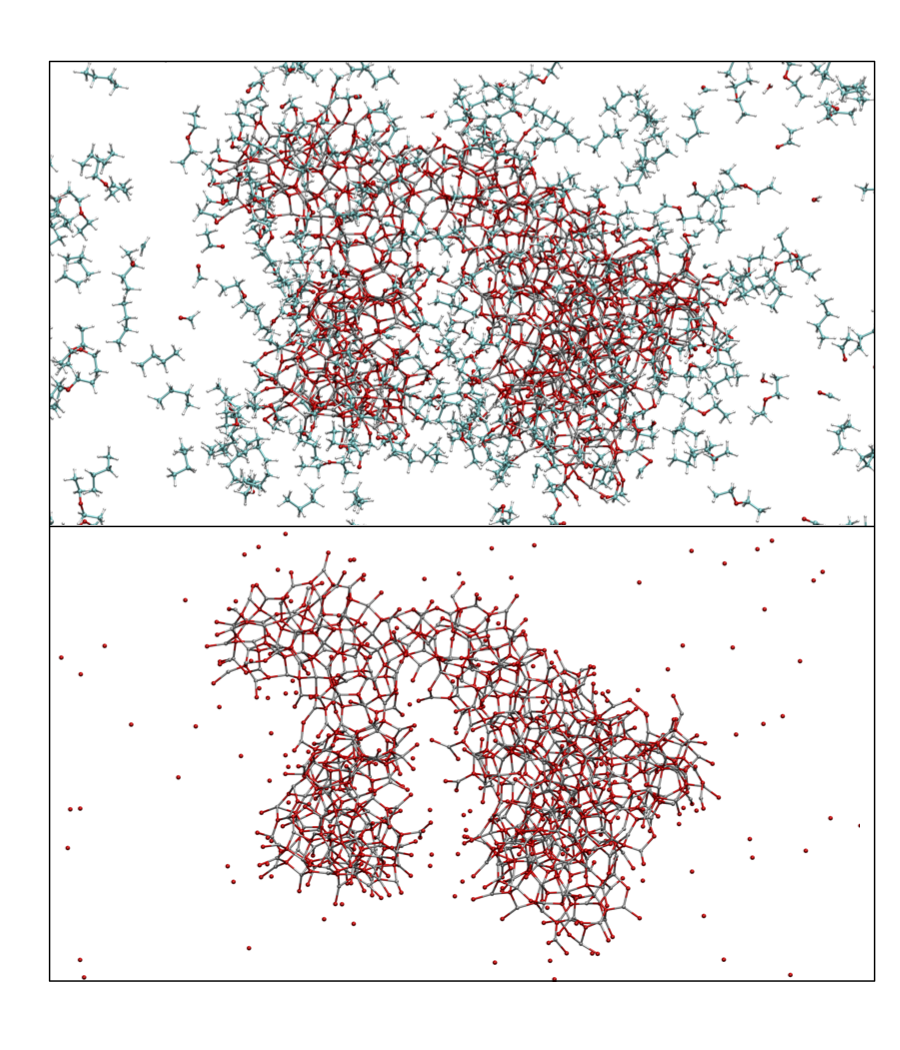
Reactive force fields, however, have been developed to allow for bond breaking and making using simple classical potential energy functions. One particular reactive force field, known as ReaxFF, was first developed in 2001 to model hydrocarbons and has more recently been used to simulate systems including explosive materials and alloys. In the Schatz group, we have have parametrized ReaxFF to model the reaction that occurs when diethyl zinc is exposed to epoxidized graphene. In experiments, zinc oxide nanoparticles form under these circumstances. As shown in Figure 1, the molecular dynamics simulations with ReaxFF successfully show how oxygen is inserted into diethyl zinc on the graphene surface. Subsequent simulations modeled the coalescence of diethoxy zinc molecules providing a structure for the nanoparticles that would form from these simulations (see Figure 2). The nanoparticle that formed did not have crystalline structure, although most of the atoms were three or four coordinate. TDDFT calculations revealed that the electronic absorption spectrum of these particles would be red-shifted by a few tenths of an eV compared to the stable wurtzite crystal phase for zinc oxide.
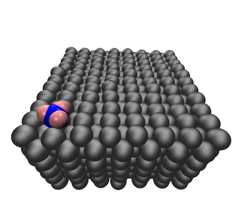
We have also applied existing ReaxFF forcefields to study reactive processes of plasma species on catalyst surfaces. The dry reforming of methane (DRM) is an attractive process due to its simultaneous consumption of two potent greenhouse gases and production of syngas, which is a crucial starting material for many value-added chemicals. The combination of a plasma discharge with a traditional DRM catalyst has been reported to demonstrate energy efficiency and product selectivity that exceed the sum of the individual processes. This effect is not well understood, and there is a lack of fundamental knowledge regarding the interaction between the plasma species and the catalyst. We utilized ReaxFF simulations to study impacts of gas phase methyl radicals on Ni[100], Ni[110], and Ni[111] surfaces at temperatures from 400 – 1600 K. The adsorption probability was relatively independent of temperature, but cleavage of a C—H bond after adsorption was most likely at temperatures above 800 K. The methyl radicals produced in the plasma discharge have a much higher adsorption probability than the methane molecules, which suggests a potential explanation for the plasma-catalyst synergy demonstrated in experiments.
Wei Lin, postdoctoral fellow
Recent Publications:
Tainter, C. J.; Schatz, G. C., “Reactive Force Field Modeling of Zinc Oxide Nanoparticle Formation”. J. Phys. Chem. C, 2015, submitted.
Johns, J.E.; Alaboson, J.M.P.; Patwardhan, S.; Ryder, C.R.; Schatz, G.C.; Hersam, M.C., “Metal Oxide Nanoparticle Growth on Graphene via Chemical Activation with Atomic Oxygen”. J. Am. Chem. Soc. 2015, 135, 18121-18125.
van Duin, A. C. T,; Dasgupta, S.; Lorant, F.; Goddard III, W. A., “ReaxFF: A Reactive Force Field for Hydrocarbons”. J. Phys. Chem. A 2001, 105, 9396-9409.
Zhang, L.; Zybin, S. V.; van Duin, A. C. T.; Dasgupta, S.; Goddard III, W. A., “Carbon Cluster Formation during Thermal Decomposition of Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine and 1,3,5-Triamino-2,4,6-trinitrobenzene High Explosives from ReaxFF Reactive Molecular Dynamics Simulations.” J. Phys. Chem. A 2009, 113, 10619-10640.
Shin, Y. K.; Kwak, H.; Zou, C.; Vasenkob, A. V.; van Duin, A. C. T., “Development and Validation of a ReaxFF Reactive Force Field for Fe/Al/Ni Alloys: Molecular Dynamics Study of Elastic Constants, Diffusion, and Segregation”. J. Phys. Chem. A 2012, 116, 12163-12174.
Johns, J. E.; Alaboson, J. M. P.; Patwardhan, S.; Ryder, C. R.; Schatz, G. C.; Hersam, M. C., “Metal Oxide Nanoparticle Growth on Graphene via Chemical Activation with Atomic Oxygen”. J. Am. Chem. Soc. 2013, 135, 18121-18125.
Mueller, J. E.; van Duin, A. C. T.; Goddard III, W. A., “Development and Validation of ReaxFF Force Field for Hydrocarbon Chemistry Catalyzed by Nickel”. J. Phys. Chem. C 2010, 114, 4939-4949.
Neyts, E. C.; Ostrikov, K.; Sunkara, M. K.; Bogaerts, A., “Plasma Catalysis: Synergistic Effects at the Nanoscale”. Chem. Rev. 2015, Article ASAP.