 Hello,
Hello,
all living organisms and man-made soft materials (from your outdoor rainjacket to the screen you are facing at the moment) are composed of thermally active molecules with well-defined functions, structures and sizes. These constituing molecules are held together by weak molecular interactions and thus can change their conformations and positions transiently to respond various external stimuli, such as electric field, flow, pH, etc. This versatility provides unprecedented survival and evolutionary capabilities in biological environment but also is a key element to design next generation active materials and smart drugs.
My research aims to understand kinetics and stimuli-responsive properties of synthetic and biological systems at molecular scales by using advanced computer simulations and analytical approaches. I employ simulation schemes both on atomistic and coarse-grained levels, as well as statistical mechanical approaches. Shoulder to shoulder with my colleagues, we try to resolve various complex problems in biology and materials sciences, on which we have currently either none or limited understanding.
Research Highlights
Co-Assembly of Peptide Amphiphiles (PAs) and Lipids into Supramolecular Nanostructures
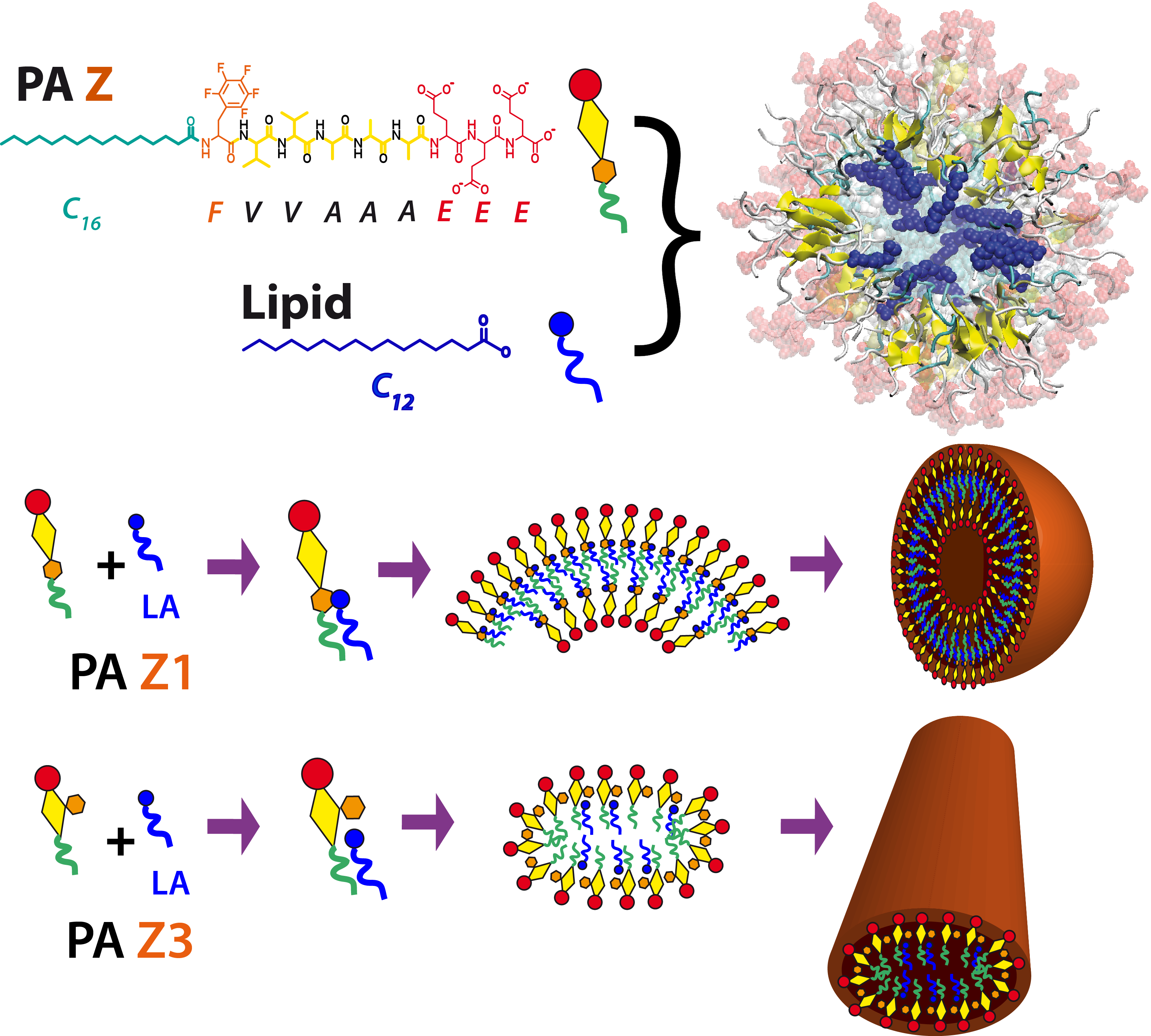
Co-assembly of binary systems driven by specific non-covalent interactions can greatly expand the structural and functional space of supramolecular nanostructures. Examples include co-assembly of bioactive and nonbioactive molecules to modify the intensity of smart drugs, or functional biomimetic structures. In our work, the self-assembly of peptide amphiphiles (PAs) and fatty acids (lipids) driven primarily by anion−π interactions are focused. With a low content of the lipid, the cylindrical nanofiber morphology is preserved. However, as the aromatic units are placed along the peptide backbone away from the hydrophobic residues, the interactions with the lipids transform the cylindrical supramolecular morphology into ribbon-like structures. Increasing the stoichiometric ratio of the lipid to PA leads to either the formation of large spherical vesicles in the binary systems or a heterogeneous mixture of assemblies. Our findings reveal that specific inter-molecular interactions can drastically change supramolecular morphology and bridge between nano to micro scales. Read more on JACS.
Facilitated dissassociation of proteins from single binding sites
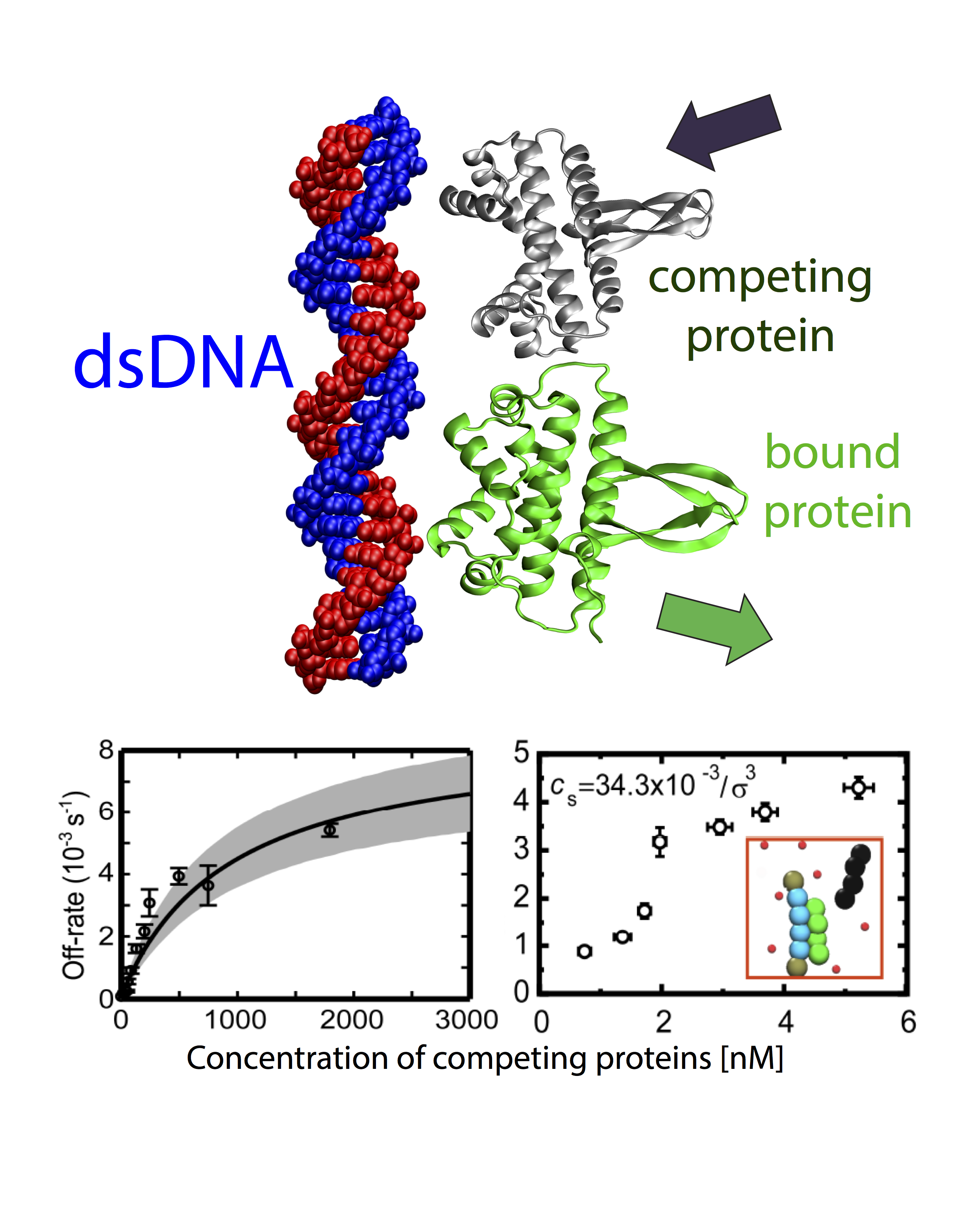 The mechanism, by which meters-long DNA is packed into micrometer-scale volumes (1 micrometer is 1000 times smaller than 1 mm) inside nuclei of mammalian cells as well as in the interior of bacterial cells, has been puzzling scientists for decades. In addition to this puzzle, DNA, whilst packed in small volumes, can simultaneously execute complex functions, such as gene regulation, protein transcription, cell division, by interacting with a complex and large array of proteins. The way proteins interact with DNA determines the biological activity of all living organisms, and inevitably any malfunction in this interaction network can lead to malevolent conditions. One set of proteins that can control the biological processes in living cells by binding and unbinding to DNA are transcription factors (TFs). TFs are key players in transcription of genetic information from DNA to mRNA to produce proteins or other types of RNA. According to the standard picture of bimolecular interactions, the dwelling time of TFs on DNA molecules in cells is independent of their concentration in solution. To understand the detailed mechanism underlying FD, we designed and performed single-molecule experiments and simulations, where we measured dissociation kinetics of Fis (factor for inversion stimulator) –- a key TF in E. coli bacteria and also major bacterial nucleoid protein — from single dsDNA binding sites. We find that the lifetimes (i.e., inverse off-rates of disassociation events) saturate at large protein concentrations in solution. While spontaneous (i.e., no unbound proteins in solution) dissociation shows a strong salt dependence, we find that facilitated dissociation depends only weakly on salt. A theoretical model and molecular dynamics simulations quantitatively explain our findings successfully and show the prevalence of this phenomenon. Our results suggest FD could have a profound effect on the dynamics of biological processes that depend on the binding of proteins in vivo. Cellular gene expression profiles and protein concentration levels occurring in complex regulatory networks should be affected by FD through its ability to shorten the residency time of a wide class of proteins, which control these networks. FD may also play a role in regulating the dynamics of chromatin 3D structure, which exhibits disorders in most cancer types and terminal diseases, by facilitating the exchange of structural proteins such as histones and nucleosomes from DNA. FD of proteins may thus be an important general effect to model gene expression in living cells. Read more on PNAS.
The mechanism, by which meters-long DNA is packed into micrometer-scale volumes (1 micrometer is 1000 times smaller than 1 mm) inside nuclei of mammalian cells as well as in the interior of bacterial cells, has been puzzling scientists for decades. In addition to this puzzle, DNA, whilst packed in small volumes, can simultaneously execute complex functions, such as gene regulation, protein transcription, cell division, by interacting with a complex and large array of proteins. The way proteins interact with DNA determines the biological activity of all living organisms, and inevitably any malfunction in this interaction network can lead to malevolent conditions. One set of proteins that can control the biological processes in living cells by binding and unbinding to DNA are transcription factors (TFs). TFs are key players in transcription of genetic information from DNA to mRNA to produce proteins or other types of RNA. According to the standard picture of bimolecular interactions, the dwelling time of TFs on DNA molecules in cells is independent of their concentration in solution. To understand the detailed mechanism underlying FD, we designed and performed single-molecule experiments and simulations, where we measured dissociation kinetics of Fis (factor for inversion stimulator) –- a key TF in E. coli bacteria and also major bacterial nucleoid protein — from single dsDNA binding sites. We find that the lifetimes (i.e., inverse off-rates of disassociation events) saturate at large protein concentrations in solution. While spontaneous (i.e., no unbound proteins in solution) dissociation shows a strong salt dependence, we find that facilitated dissociation depends only weakly on salt. A theoretical model and molecular dynamics simulations quantitatively explain our findings successfully and show the prevalence of this phenomenon. Our results suggest FD could have a profound effect on the dynamics of biological processes that depend on the binding of proteins in vivo. Cellular gene expression profiles and protein concentration levels occurring in complex regulatory networks should be affected by FD through its ability to shorten the residency time of a wide class of proteins, which control these networks. FD may also play a role in regulating the dynamics of chromatin 3D structure, which exhibits disorders in most cancer types and terminal diseases, by facilitating the exchange of structural proteins such as histones and nucleosomes from DNA. FD of proteins may thus be an important general effect to model gene expression in living cells. Read more on PNAS.
Interactions between charged polymeric surfaces
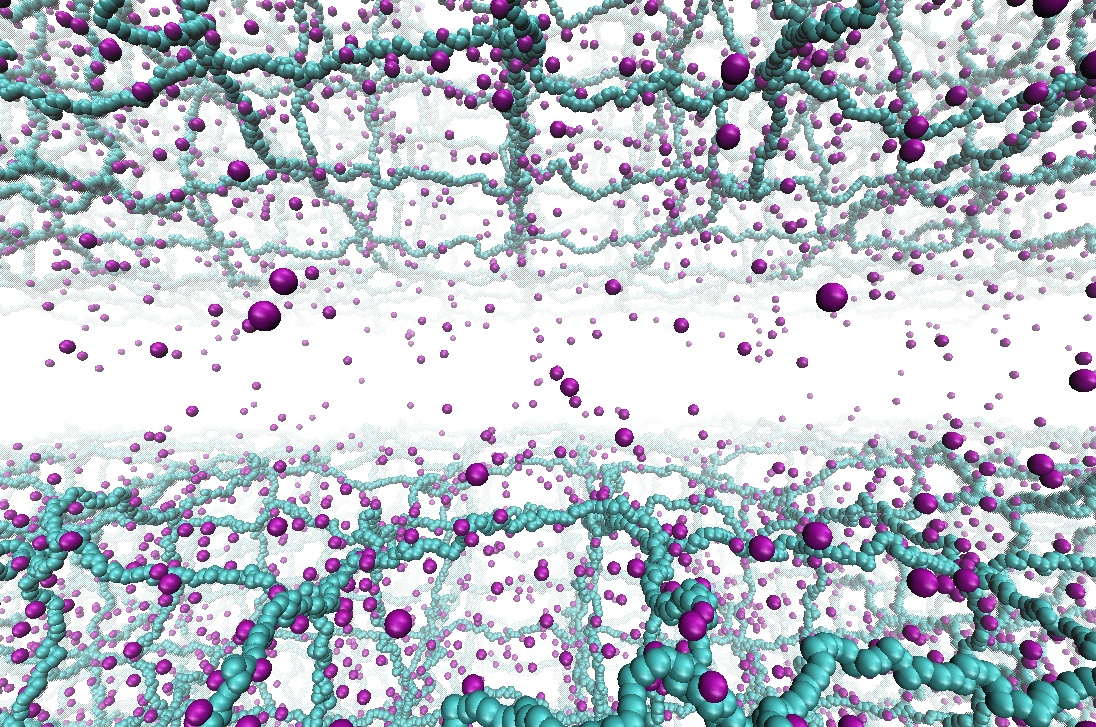
Hydrogels are highly-swollen networks of cross-linked polymers with ionizable groups. Surfaces formed by these charged-polymers (polyelectrolytes) are highly abundant in both synthetic and biological systems. These charged-polymer networks are the main constituents of contact lenses and adsorbing components of diapers, and also appear in various pharmaceutical applications in the forms of smart drugs due to their pH response. In biological machinery, hydrogels composed of biopolymers appear quite often as well, particularly in connective and inter-cellular tissues: cartilage tissue and its components (synovial joints), mucin bilayers bridging the cornea stroma and epithelial surfaces in eye, mucin-brush layers coating the interior surfaces of mammalian respiratory system (lungs) are among some examples. It is, hence, desirable to understand the interactions between the hydrogel surfaces and possibly relate experimentally measurable properties (e.g., swelling ratio or lubrication properties) to the molecular details. In our recent publication, we first revisit a previous theory based on the solution of the Poisson-Boltzmann equation and compare the results to our molecular dynamics simulations of two highly-charged hydrogel surfaces. We also introduce scaling arguments to relate electrostatic strength of solvent (also referred as ionic strength) and polymerization degree to the observed pressure regimes and the threshold pressure separating these regimes. Read more on Macromolecules.
Ionic transport in polyelectrolyte networks
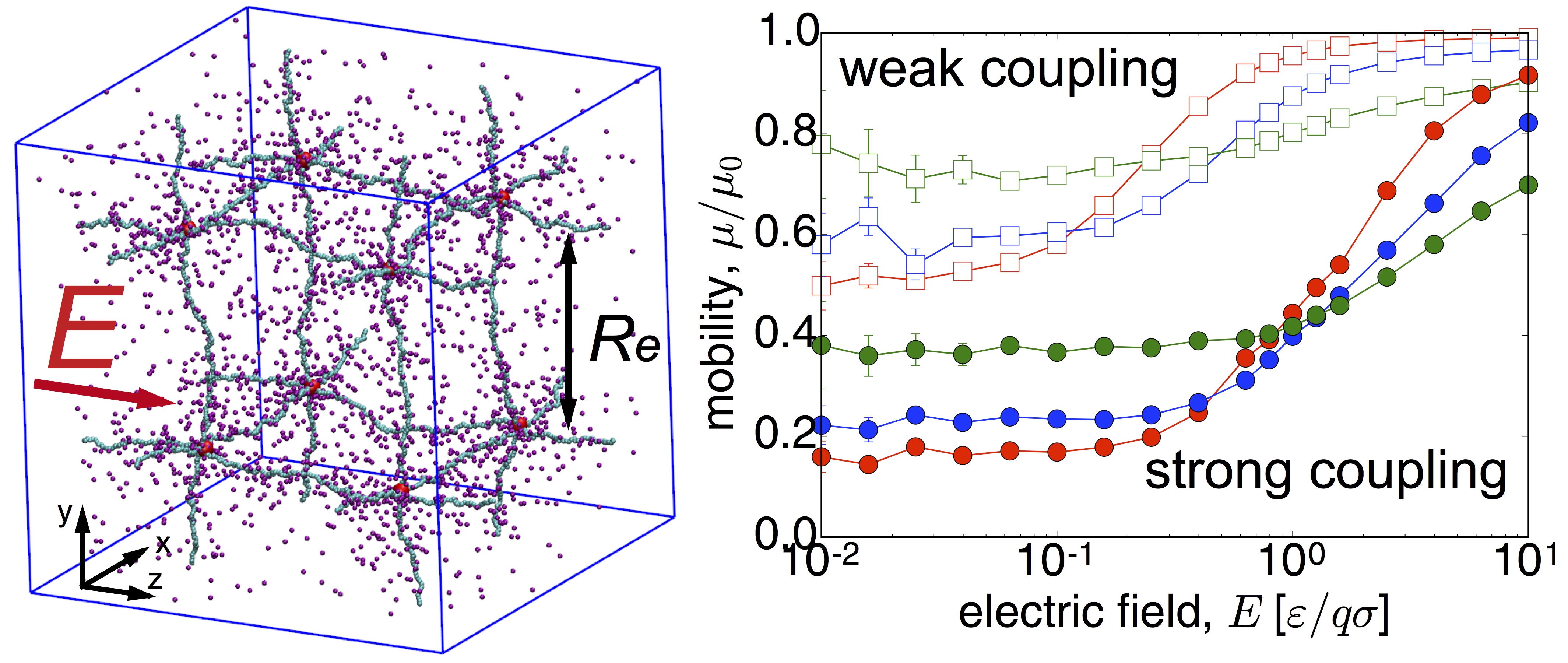
Ionic transport in heterogeneous media on the microscale and nanoscale is of paramount interest for many applications including water desalination, biomimetic systems, as well as the potential for future generations of man- made devices, which can interface and be intrinsically compatible with biological tissues. Coarse-grained molecular dynamics simulations suggest that a nonlinear response of the ionic conductivity to an applied electric field, for field strengths that are comparable to the ionic coupling strengths. This behavior correlates to a broadening of the ionic distribution around the polymer backbone under an increasing electric field. Also, we find that the weak-field ionic mobility in gels increases with density, which is opposite to the behavior of simple electrolytes. This relates to the mean coupling between charges that decreases in gels, but increases in simple elec-trolytes, with increasing density. These results provide more insights into the electric response of polyelectrolyte gels, to support the develop- ment of applications that combines electric and mechanical properties of polyelectrolyte gels for energy storage, sensing, selective transport, and signal transfer. Read more on Macromolecues .
Formation of ion-rich phases in the melts of amphiphilic molecules.
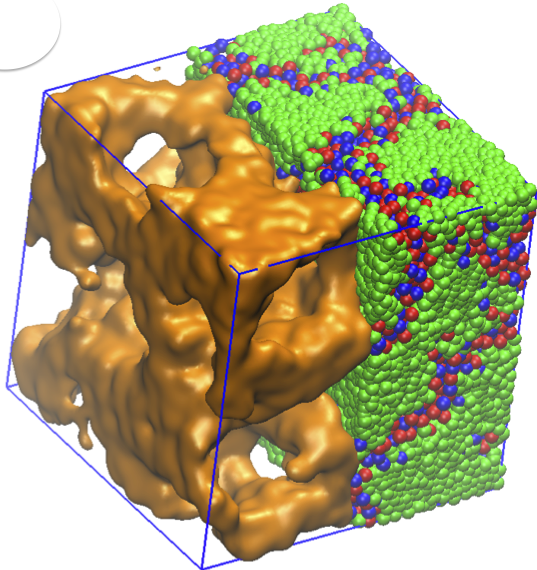 Amphiphilic molecules bear both hydrophobic and hydrophilic chemical groups. At high volume fractions, these molecules arrange themselves relative to each other in order to minimize their free energy, and form phases. Ionic liquids – with aliphatic tails and cationic groups – exhibit fascinating nanoscale morphological phases and are promising materials for energy storage applications. Liquid crystalline order emerges in ionic liquids with specific chemical structures. In our study, we investigate the phase behaviour and related ionic conductivities of dry (solvent free) ionic liquids, using extensive molecular dynamics simulations. Temperature dependence, properties of polymeric tail and excluded volume symmetry of the amphiphilic ionic liquid molecules are investigated in large scale systems with both short and long-range Coulomb interactions. Our results suggest that by adjusting stiffness and steric interactions of the amphiphilic molecules, lamellar or 3D continuous phases result in these molecular salts. We also demonstrate that, in the absence of electrostatic interactions, the morphology is distorted. Our findings inspire new design principles for room temperature ionic liquids and help explain previously-reported experimental data. Read more on PCCP .
Amphiphilic molecules bear both hydrophobic and hydrophilic chemical groups. At high volume fractions, these molecules arrange themselves relative to each other in order to minimize their free energy, and form phases. Ionic liquids – with aliphatic tails and cationic groups – exhibit fascinating nanoscale morphological phases and are promising materials for energy storage applications. Liquid crystalline order emerges in ionic liquids with specific chemical structures. In our study, we investigate the phase behaviour and related ionic conductivities of dry (solvent free) ionic liquids, using extensive molecular dynamics simulations. Temperature dependence, properties of polymeric tail and excluded volume symmetry of the amphiphilic ionic liquid molecules are investigated in large scale systems with both short and long-range Coulomb interactions. Our results suggest that by adjusting stiffness and steric interactions of the amphiphilic molecules, lamellar or 3D continuous phases result in these molecular salts. We also demonstrate that, in the absence of electrostatic interactions, the morphology is distorted. Our findings inspire new design principles for room temperature ionic liquids and help explain previously-reported experimental data. Read more on PCCP .
Electrostatic energy conversion with polyelectrolyte gels
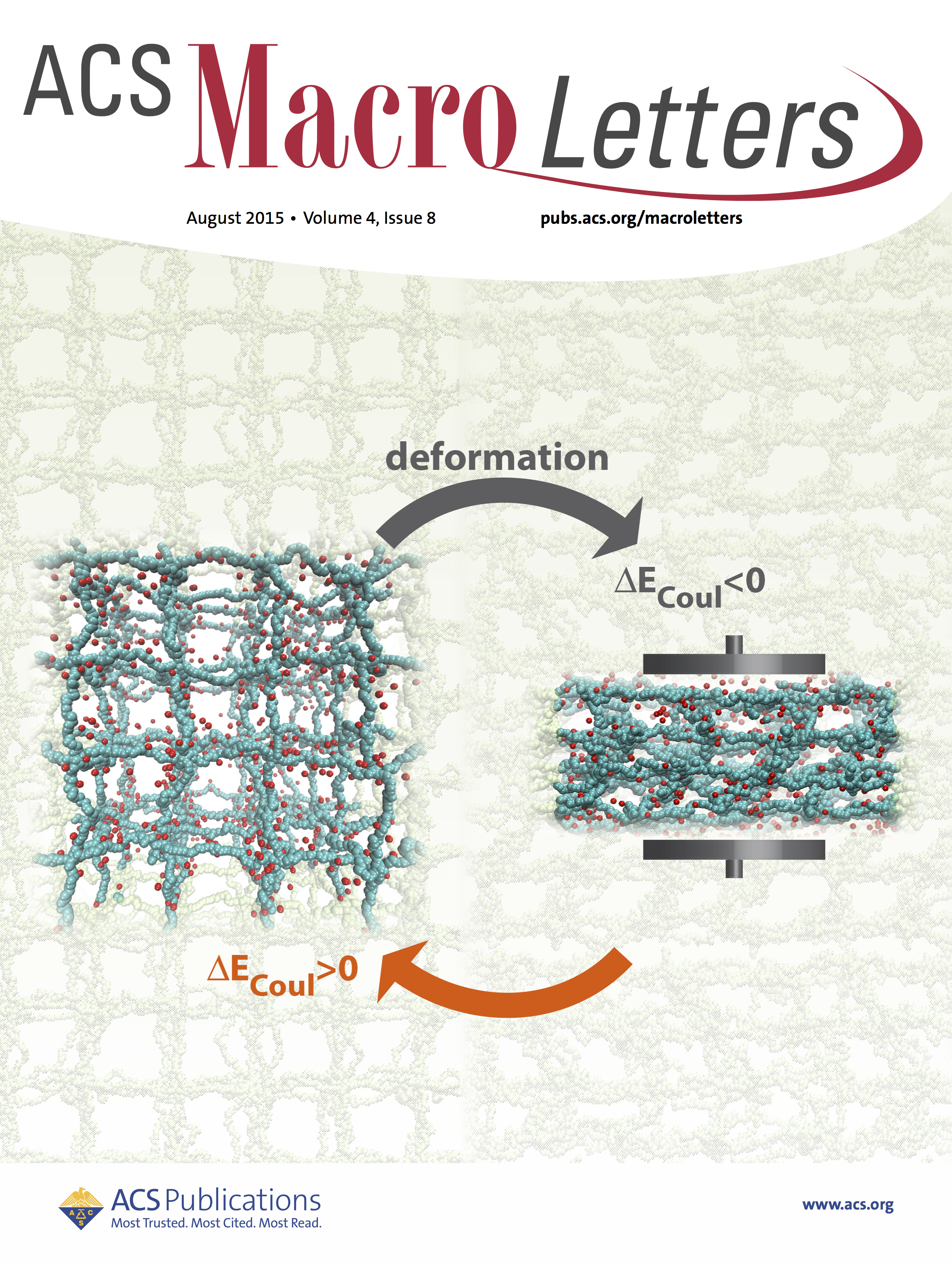 Apart, from being the main constituent of contact lenses and pill capsules, hyrogels have also proven themselves pretty effective in other applications including as mechano-electric energy converters or bio-mimetic structures. The hydrogels own their special stimuli responsive properties to their inherent electrostatic and elastic properties. Their electrostatic nature allows them to adjust their internal interactions reversibly upon application of external stimulus such as electromagnetic fields or mechanical deformations. Our MD simulations with explicit counterions showed that if a polyelectrolyte gel undergoes strain-control deformations, applied mechanical energy is mainly converted into electrostatic energy rather than other energy components (i.e., elastic and steric energies). The energetic change is due the decreasing translational entropy of counterion gas trapped inside the gel in combination with chain elasticity of PE gels. This scenario is only possible if ionized counterions cannot vacate the gel, which is ensured by the local electro-neutrality condition. Read more on ACS Macro Letters.
Apart, from being the main constituent of contact lenses and pill capsules, hyrogels have also proven themselves pretty effective in other applications including as mechano-electric energy converters or bio-mimetic structures. The hydrogels own their special stimuli responsive properties to their inherent electrostatic and elastic properties. Their electrostatic nature allows them to adjust their internal interactions reversibly upon application of external stimulus such as electromagnetic fields or mechanical deformations. Our MD simulations with explicit counterions showed that if a polyelectrolyte gel undergoes strain-control deformations, applied mechanical energy is mainly converted into electrostatic energy rather than other energy components (i.e., elastic and steric energies). The energetic change is due the decreasing translational entropy of counterion gas trapped inside the gel in combination with chain elasticity of PE gels. This scenario is only possible if ionized counterions cannot vacate the gel, which is ensured by the local electro-neutrality condition. Read more on ACS Macro Letters.